
米国医学研究者のDr. Pontaです。今回はアメリカの医学研究ネットワークThe Cancer Genome Atlas(TCGA) Programにより発表された各悪性腫瘍のマルチオミクスデータのダウンロード方法について、解説します。
TCGAbiolinksとは
TCGAで発表されたデータはNIHで管理さているGDCデータポータルに保存されており、BamやVCFファイルを除き公共公開されています。BamやVCFは研究計画などの登録申請、審査を経てアクセスできるようになります。また各患者ごとのにファイルが公開されているので、コホートレベルの解析を実施するにはファイルをそれぞれダウンロードしなければならないので、手間と時間がかかります。
このような状況下で、臨床的に有用な公開データセットを簡単に扱えるようにしたRパッケージが”TCGAbiolinks”です。 他にもいくつかTCGAデータを扱うパッケージがありますが、これが最もメジャーと思います。では使い方をさっそく見ていきましょう。
使用方法
インストール
TCGAbiolinksはBiocManager経由でインストールします。
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("TCGAbiolinks")登録データセットの確認
# ライブラリの読み込み
library(TCGAbiolinks)
# 登録データセット情報の取得
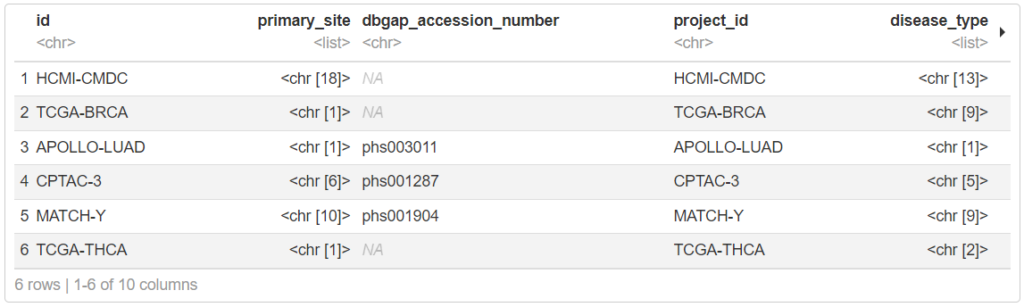
projects <- getGDCprojects()
head(projects)一覧の通り、TCGA以外のデータセットも追加登録されています。
データセットの内容確認
例としてTCGA-LUAD(肺腺癌コホート)のデータとしてどのようなデータがあるかをみてみます。
getProjectSummary("TCGA-LUAD")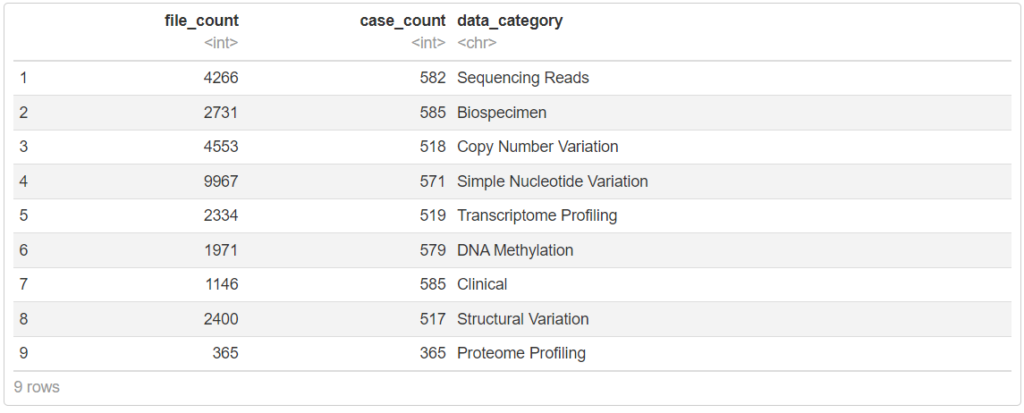
500症例以上のCNVやSNV、トランスクリプトーム情報、臨床情報が登録されていることがわかります。
TCGA-LUADデータのダウンロード
ここでは例としてSNV(変異データ)として”Gene Expression Quantification”、”Isoform Expression Quantification”、”miRNA Expression Quantification”の3つがあります。”Gene Expression Quantification”がいわゆる一般に使われるRNAseqデータで、workflow.typeとして”STAR – Counts”、”HTSeq – FPKM-UQ”、”HTSeq – FPKM”が登録されています。
ダウンストリームでDEseq2, edgeR, limmaなどを用いて発現差解析を実施する場合はインプットがカウントデータなので、Countsを選択してダウンロードします。
SNV
SNVとしてはヒトゲノムリファレンスhg19とhg38にマッピングされた二種類があります。hg38がより最新のヒトゲノム配列なので、hg38を今回はダウンロードします。
# クエリを作成
query <- GDCquery(
project = "TCGA-LUAD",
data.category = "Simple Nucleotide Variation",
access = "open",
data.type = "Masked Somatic Mutation",
workflow.type = "Aliquot Ensemble Somatic Variant Merging and Masking"
)
# ダウンロード
GDCdownload(query)
# ダウンロードしたファイルをオーガナイズし、読み込む
maf <- GDCprepare(query)
# Rオブジェクトで保存
save(maf, file = "data/TCGA_LUAD_SNVmaf.RData")MAFファイルに格納されたSNV情報はRパッケージ”maftools”で簡単にサマリーを作成できます。
library(maftools)
maf1 = read.maf(maf = maf)
# MAFサマリ作成
plotmafSummary(maf = maf1, rmOutlier = TRUE, addStat = 'median', dashboard = TRUE, titvRaw = FALSE)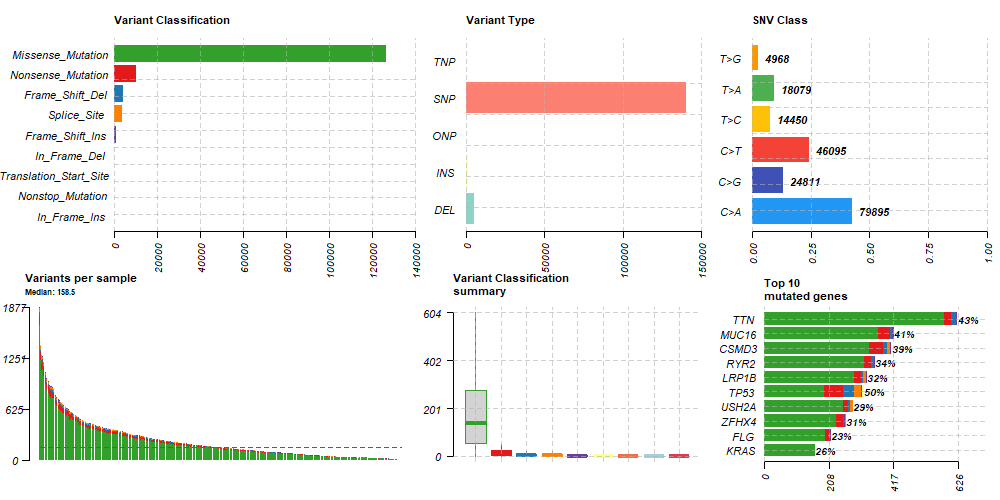
オンコプリントも簡単に作成できます。
oncoplot(maf = maf1, top = 10)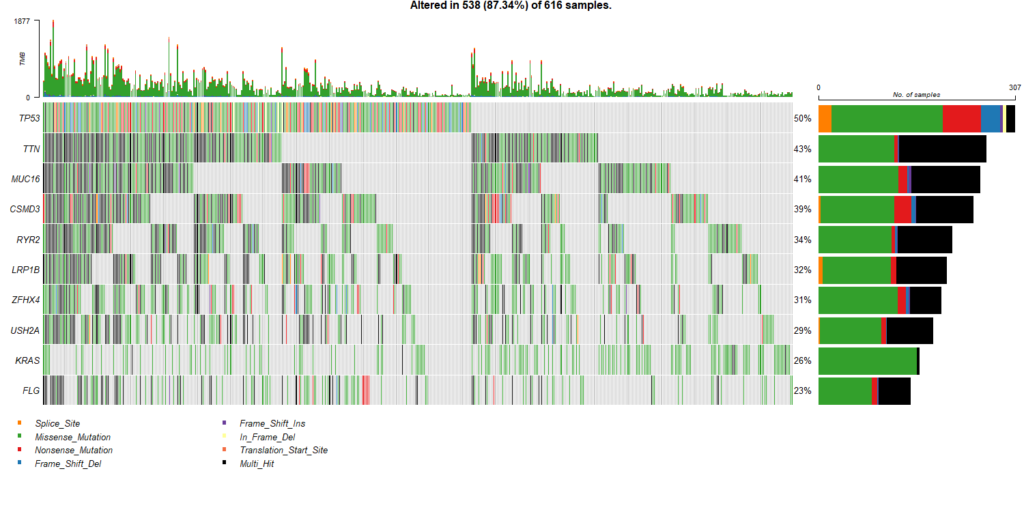
トランスクリプトームデータ
RNAseqのリードカウントデータ(STARパッケージ:シーケンスリードのマッピング、カウントツール)は以下のようにしてダウンロードします。
# クエリ作成
query <- GDCquery(project = "TCGA-LUAD",
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "STAR - Counts")
# ダウンロード
GDCdownload(query)
# データ読み込み(S4形式)
data <- GDCprepare(query)
# S4データフォーマットからカウントデータを取得
# 60660の遺伝子名は行に入力されている
library(data.table)
exp <- data.table(assay(data)[1:60660,],rownames = TRUE)
# S4データフォーマットからENSEMBL_ID, HUGO gene symbolを取得
exp$gene_id <- data@rowRanges$gene_id
exp$gene_name <- data@rowRanges$gene_name
# 使いやすいように遺伝子名を1、2例目に移動し、データ確認
exp <- exp[, c(602,603, 1:601)]
head(exp[,1:5])
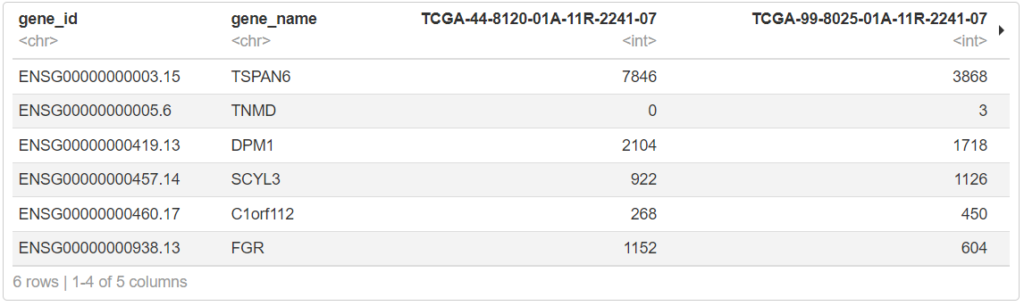
# Rオブジェクトで保存
save(exp, file = "data/TCGA_LUAD_exp.RData")臨床情報
臨床情報はシンプルに以下のコードでダウンロード、保存できます。
# コホート名を指定し、臨床情報をダウンロード
clinical <- GDCquery_clinic("TCGA-LUAD")
head(clinical)
# Rオブジェクトで保存
save(clinical, file = "data/TCGA_LUAD_clinical.RData")
まとめ
以上のように、数十分で500症例以上のSNVファイル、mRNA発現値データ、基本的な臨床情報を簡単にダウンローすることができます。データをよくまとまった形でダウンロードできるのはダウンストリーム解析を容易にしますので、大変重要なポイントです。自分の研究データの裏付けを外部の実臨床データでValidationしたいときには、TCGAデータセットは貴重なリソースなので、是非使いこなせるようになることをお勧めします。

