
米国医学研究者のDr. Pontaです。
今回はシグナルパスウェイのエンリッチメント解析方法について解説します。パスウェイ解析を行う方法は、MetascapeやiDEPのようにWebベースのものや、Rの”clusterProfiler”のようにローカル環境で行う方法があります。私の経験上、”clusterProfiler”がもっともカスタム性が高く、使い勝手がよいので今回はこちらを使用して、説明します。
はじめに
基本の確認ですが、パスウェイ解析では、発現変動のあった遺伝子群がどのシグナルパスウェイに多く含まれていたかを統計的に検定します。この解析により、発現変動遺伝子群にどのような既知のパスウェイがエンリッチしていたかが、わかります。パスウェイ解析を実施するには、変動遺伝子群と既知パスウェイに所属するそれぞれの遺伝子リストが最低限必要ということになります。パスウェイ遺伝子リストあるいは生物学的機能モジュールごとにまとめられた遺伝子リストとして、GO term、KEGG Pathways、Reactome、MsigDBがあります。
Gene Set Enrichment Analysis
GSEAとはGene Set Enrichment Analysisの略です。任意の二群間比較における遺伝子発現差を入力とし、累積分布関数を用いて、エンリッチメントスコアと呼ばれる尺度を計算します。このスコアがエンリッチメントの程度を示し、パスウェイごとの変動の”向き”と”程度”が評価できます。
Over Representative Analysis
Over representative analysis (ORA)では、遺伝子名のみを入力としており、GSEAと違い発現差情報は解析に用いません。そのため、ORAではエンリッチしているか否かしか基本的に述べることができません。
MSigDB(Molecular Signatures Database)
MSigDBは、遺伝子発現データに関連する遺伝子セット(gene sets)の大規模なコレクションを提供する公共のデータベースです。これは、遺伝子やタンパク質の活動を特定の生物学的状態や疾患に関連付けるために使用されます。Broad Instituteなどによって運営されており、研究者や生命科学関係者による無料のオンラインリソースとして利用できます。こちらからダウンロードできます。
解析実践
エンリッチメント解析のためのパッケージインストール
clusterProfilerはBioconductorで維持管理されているので、以下のようにしてローカルのR環境にインストールします。同時にヒト遺伝子名を扱うため、org.Hs.eg.dbパッケージも同様にインストールします。
if (!require(“BiocManager”, quietly = TRUE)) install.packages(“BiocManager”)
BiocManager::install(“clusterProfiler”)
if (!require(“BiocManager”, quietly = TRUE)) install.packages(“BiocManager”)
BiocManager::install(“org.Hs.eg.db”)模擬データの準備
今回はTCGA胃がんにおけるMSIタイプとGSタイプのmRNA発現差解析結果をダウンロードして、テストデータとします。以下のようにcBioportalのSTADデータセットにおいて、GS型とMSI型のmRNA発現差解析ページにアクセスし、解析結果をテキストデータとしてダウンロードします。以下のパスウェイ解析では、このデータを使っていきます。cBioportalの使い方はこちらで解説してますので、よろしければ一緒にご覧ください。
R環境でのデータ読み込み、整形
ここからRstudio内での作業になります。clusterProfilerのインプットはベクトル形式なので、以下のようにダウンロードしてきた発現差解析結果をもとに、ベクトル入力データを作成します。
# 必要パッケージの読み込み
library(clusterProfiler)
library(org.Hs.eg.db)
library(tidyverse)
library(enrichplot)
# ダウンロードしたデータの読み込み
d <- read.delim("data/TCGA_Stomach_GSvsMSI_table.tsv")
# gene, log2 fold change, qvalueを抜き出し、列名変更
d <- d[c(1,7,9)]
colnames(d) <- c("Gene", "Log2FC", "Qvalue")
# 統計的に有意な遺伝子のみ選択
d <- d %>% filter(Qvalue < 0.05)
# 念のため、ダブリ除去処理をする
d <- d[!duplicated(d$Gene),]
# 遺伝子シンボルからENTREZID情報を取得し、元のデータフレームに加える
eg = bitr(d$Gene, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
d <- d %>% filter(d$Gene %in% eg$SYMBOL)
d <- merge(d, eg, by.x="Gene", by.y="SYMBOL")
# 発現値を降順に並べたベクトルをつくり、各要素名としてENTREZIDをいれる
geneList <- d[,c("Log2FC")]
names(geneList) <- as.character(d[,c("ENTREZID")])
geneList <- sort(geneList, decreasing =TRUE)
# Log2FCの絶対値が2より大きいものの遺伝子名ベクターを作成
gene <- names(geneList)[abs(geneList) > 2] # hold only gene id以上のコードで、GSEAに必要なランク付き遺伝子名データ(上記コードで”geneList”)とORAに使う(ランク情報なしの)遺伝子名データ(上記コードで”gene”)が準備できました。
GO エンリッチメント解析
では早速GOBPエンリッチメント解析をやってみます。
# Overrepresentative test for GO-BP
ego <- enrichGO(gene = gene,
universe = names(geneList),
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05,
readable = TRUE)
# GSEA test for GO-BP
ego2 <- gseGO(geneList = geneList,
OrgDb = org.Hs.eg.db,
ont = "BP",
minGSSize = 20,
maxGSSize = 500,
pvalueCutoff = 0.05,
verbose = FALSE)次にORAの結果をグラフ化します。
# Barplot of ORA test of GO_BP result、下左図
barplot(ego, showCategory=5) + ggtitle("GO_BP over representative analysis")
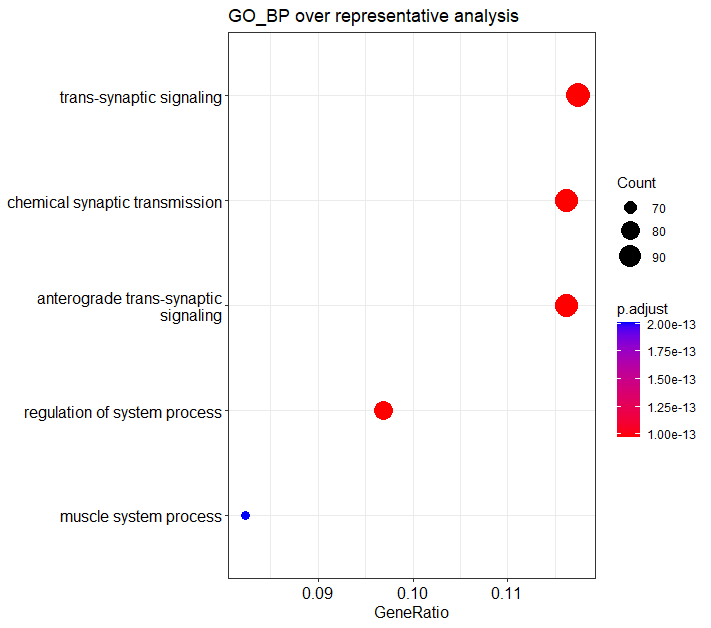
# Dotplot of ORA test of GO_BP result、下右図
dotplot(ego, showCategory=5) + ggtitle("GO_BP over representative analysis")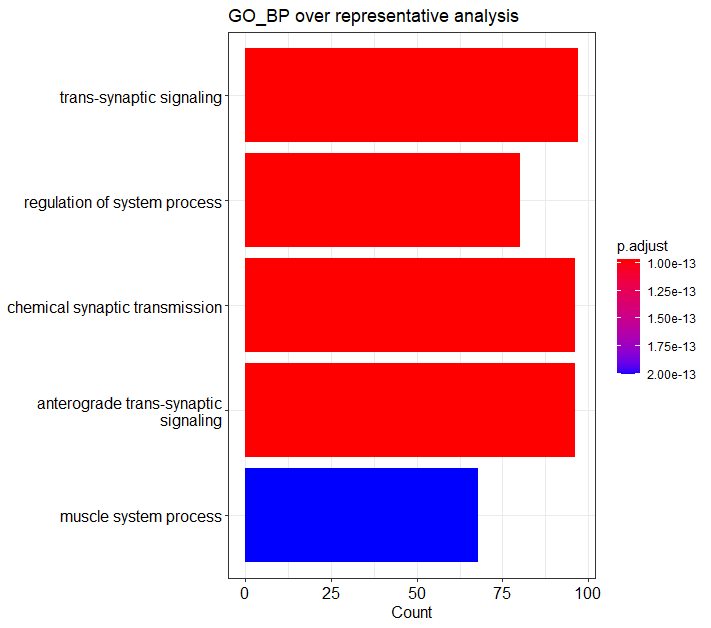
次にGSEAの結果をリッジプロットでグラフ化します。
ridgeplot(ego2, showCategory = 5) + ggtitle("GO_BP GSE analysis")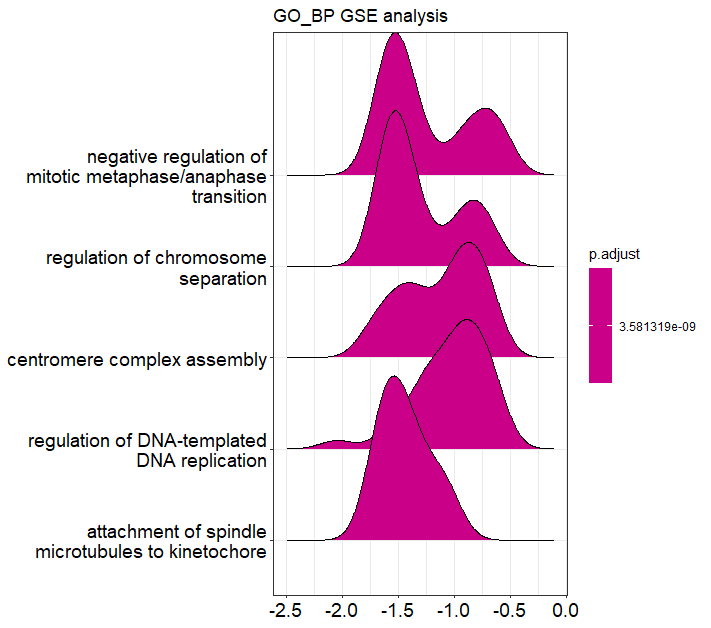
GSEAでは統計的有意にエンリッチしていたTermがスコア上で”多い”のか”少ない”のかがわかります。つまりその経路・プロセスが対象群で亢進しているのか、抑制されているのかが大まかにスコア値としてわかります。これがORAとのもっとも大きな違いです。
KEGGエンリッチメント解析
KEGGについても同様にORA、GSEA解析ができます。
# KEGG ORA test
kk <- enrichKEGG(gene = gene,
organism = 'hsa',
pvalueCutoff = 0.05)
# KEGG GSEA test
kk2 <- gseKEGG(geneList = geneList,
organism = 'hsa',
minGSSize = 20,
pvalueCutoff = 0.05,
verbose = FALSE)
KEGGのGSEA結果を2つの方法でグラフ化します。
# リッジプロットで統計的有意な経路、TOP10を表示(QまたはPvalueに基づきTOP10が自動セレクトされる)(下左図)
ridgeplot(kk2, showCategory=10)
# UPとDownのそれぞれTOP5を表示するには以下コード(下右図)
y <- arrange(kk2, -abs(NES)) %>% group_by(sign(NES)) %>% dplyr::slice(1:5)
ggplot(y, aes(NES, fct_reorder(Description, NES), fill=qvalue), showCategory=10) +
geom_col(orientation='y') +
scale_fill_continuous(low='red', high='blue', guide=guide_colorbar(reverse=TRUE)) +
theme_minimal()+ ylab(NULL)+ggtitle("KEGG GSE analysis")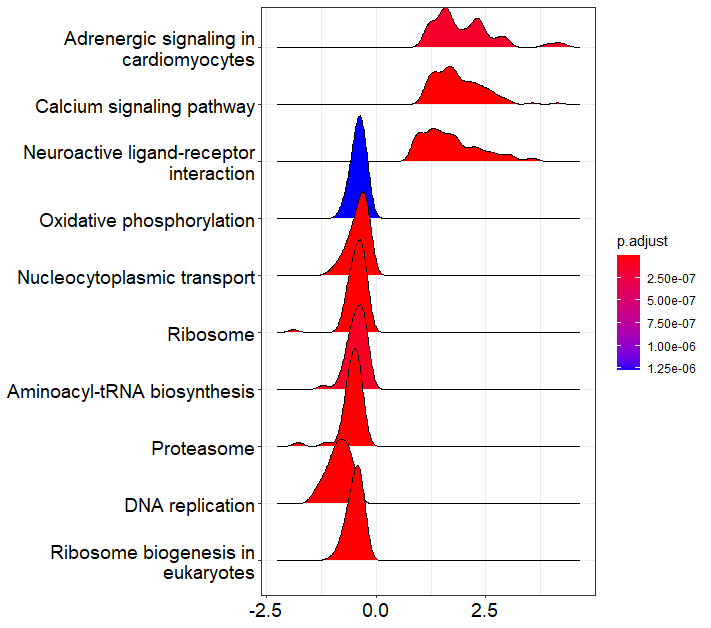
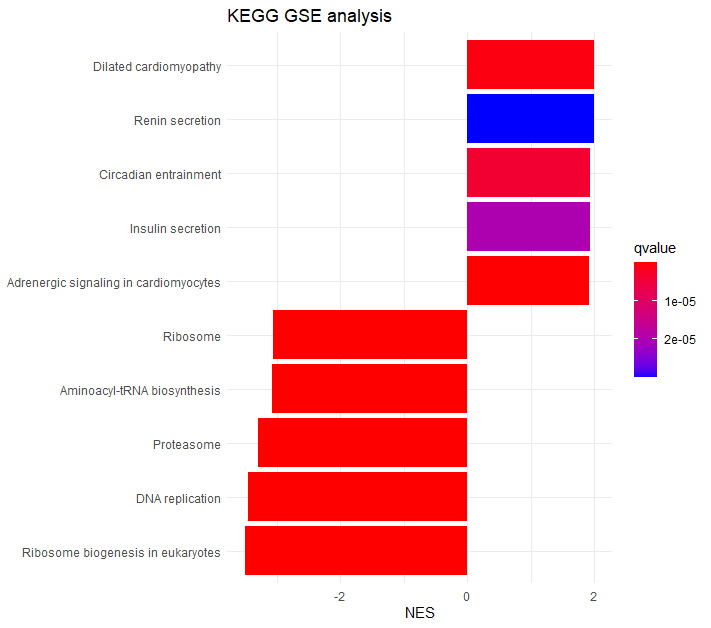
右図のほうが、一枚の図でUPとDownを同時に表示できるので、多くの情報をなるべく小さく要約する必要のある科学論文では有用と思います。実際筆者もこちらの図を論文では多用しています。
MSigDBを使用したエンリッチメント解析
がん生物学系の論文を読んでいるとHALLMARKという遺伝子セットを一度は目にしたことがあるのではないでしょうか。今回はどのようなHallmark遺伝子セットが胃がんの発現差結果にエンリッチしているかを解析してみます。
# Hallmark遺伝子セットを読み込む
H <- read.gmt("data/msigdb_v2023.1.Hs_GMTs/h.all.v2023.1.Hs.entrez.gmt")
# ORA実施
egmt_H <- enricher(gene, TERM2GENE=H)
# GSEA実施
egmt2_H <- GSEA(geneList, minGSSize = 20,TERM2GENE=H, verbose=FALSE)以下でGSEA結果のみ確認します。
# GSEA result TOP10のUP,DOWNを表示
y <- arrange(egmt2_H, -abs(NES)) %>% group_by(sign(NES)) %>% dplyr::slice(1:10)
ggplot(y, aes(NES, fct_reorder(Description, NES), fill=qvalue), showCategory=20) +
geom_col(orientation='y') +
scale_fill_continuous(low='red', high='blue', guide=guide_colorbar(reverse=TRUE)) +
theme_minimal()+ ylab(NULL)+ggtitle("MsigDB Hallmark GSE analysis")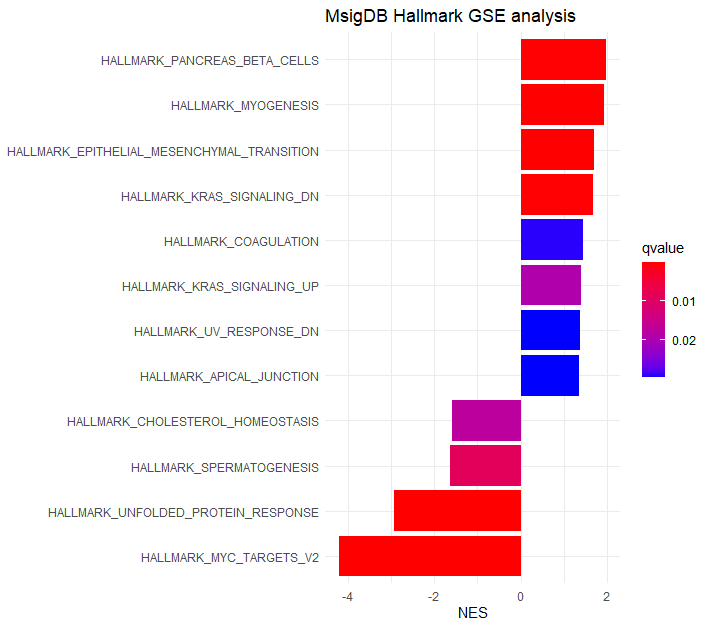
HALLMARK遺伝子セットとしては、MSI型に対しGS型ではEMTなどの高悪性度癌の特徴がエンリッチしてるようです。GS型は組織型としてびまん型胃がんが主なので、既存の臨床データとしても合致しており、解析結果に納得できます。
GSEAプロットの作り方
GSEA結果に基づき、GSEAプロットを作りたいときは以下のようにします。
ここでは以下の三つを表示することとします。”HALLMARK_MYOGENESIS”、”HALLMARK_APICAL_JUNCTION”、”HALLMARK_MYC_TARGETS_V2″
# 表示したいgeneSetIDを調べる
which(rownames(egmt2_H@result)=="HALLMARK_MYOGENESIS") # ID=3
which(rownames(egmt2_H@result)=="HALLMARK_APICAL_JUNCTION") # ID=12
which(rownames(egmt2_H@result)=="HALLMARK_MYC_TARGETS_V2") # ID=1
# 調べたgeneSetIDを以下のごとく入力し、作図
gseaplot2(egmt2_H, geneSetID = c(3,12,1), base_size = 11, subplots = 1:2,rel_heights = c(1, 0.3))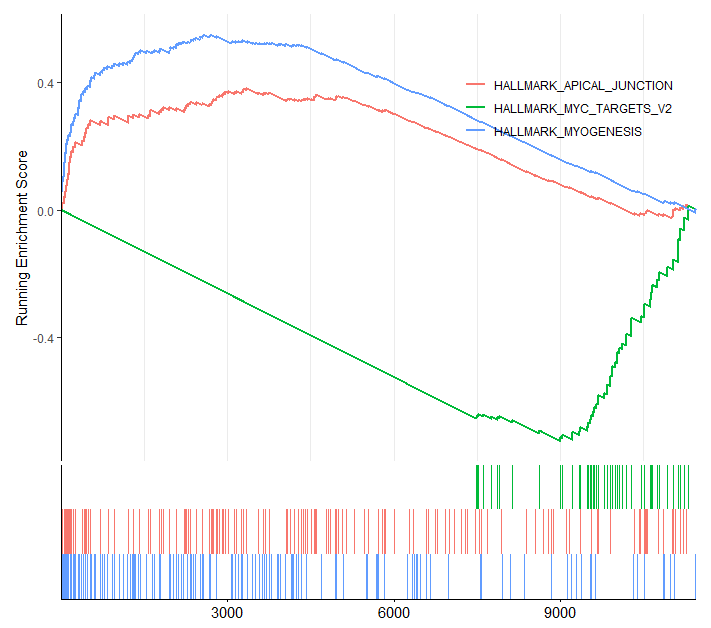
特定のパスウェイについて結果を表示したいときは、こちらのGSEAプロットのほうが情報量が多く、論文でも好まれていると思います。通常この図にNSE値やPvalueも手動で追加して最終論文図として使っています。
まとめ
以上、clusterProfilerの基本的な使用説明でした。一度コードを書いてしまえば、解析条件が変わって新しい発現差結果が出てきても、入力データを置き換えるだけで済みます。多量の発現データがあり、さまざまなエンリッチメント解析を実施する必要があっても、簡単にミスなく処理できます。コードも保存されるので、後日どんなエンリッチメント解析を実施したかレビューするときも問題ありません。WebベースサービスのMetascapeなどは繰り返し処理や後日のレビューし易さといった点でRstudioのローカル解析に劣ります。読者のみなさんはどうでしょうか。実際に使用してみると、ClusterProfilerの多くの利点にお気づきになるのではないかと思います。
今回も最後までお読みいただき、ありがとうございました。
