
米国医学研究者のDr. Pontaです。
今回は高次元データ解析時(RNA-seqなどの数万次元からなる行列など)の主成分分析(PCA)について解説します。
PCAとは
PCAはデータの次元削減と特徴量抽出のために使用される統計的手法です。PCAでは高次元のデータを相対的に低次元の空間に射影することで、データの構造やパターンを把握しやすくすることを目的としています。具体的には多変量データセットの特徴間の相関関係を分析し、それらの相関関係を最もよく表現する少数の主成分(特徴量)を抽出します。これらの主成分は、データの分散が最大になるように選ばれます。主成分は、元の特徴空間の軸とは異なる新しい軸として捉えることができ、元のデータの持つ情報をできるだけ保持しつつ、次元を削減することができます。
何に役立つか?
医学データ分析でも非常によく使用される解析手法です。たとえば臨床サンプルのRNAseqの正規化後の発現データがあったとして、本番のダウンストリーム解析を行う前に、通常発現データに予期しないバイアスやバッチエフェクトがないかを確認する必要があります。例えば、臨床サンプルの保存期間や凍結するまでの処理時間などによって、データに偏りがないかをPCAや相関係数で大まかに確認します。逆に既存の研究から明らかに発現プロファイルが異なるとわかっているような因子では、その因子によってPCAでの分布が分かれることを確認することにより、その実験系が既知の違いを検出できていることを示し、サンプル処理、データ前処理が適切に行われたことをサポートするデータになります。このようなことから、大規模臨床コホートでは必ずと言っていいほど、これらのデータが論文サプリメントなどに記載されています。このように発現データなどの数値データを扱うときに必須となる数学的手法です。
使い方実践
発現データ準備
今回は誰でも簡単にできるように、cBioPortalからウェブブラウザー経由でデータをダウンロードし、Rに読み込む形でやってみます。cBioPortalの詳細はこちらに記載してありますので、よければ合わせてご覧ください。以下のようにcBioportalへアクセスし、TCGA胃がんコホートの全情報をダウンロードし、解凍、RstudioのWorking Directory下に保存します。
次に以下のようにRで読み込みデータの成型を行います。
# 発現データ読み込み(column方向にサンプルID、row方向に遺伝子名)
exp <- read.delim("data/stad_tcga_pan_can_atlas_2018/stad_tcga_pan_can_atlas_2018/data_mrna_seq_v2_rsem.txt")
# 列名の”.”を”_"(underscore)に置換
colnames(exp) <- gsub("\\.", "_", colnames(exp))
# missing valueを含む遺伝子行を取り除く(PCA解析でNAがあるとできない)
exp <- exp[complete.cases(exp[ , 2:414]),] # expの発現値カラムだけ指定
# column方向にENTREZ_ID(遺伝子)、row方向にサンプルIDとなるように転置する
exp <- exp[!duplicated(exp$Entrez_Gene_Id),] # 行名はユニークである必要があり、ダブリを除去
rownames(exp) <- exp$Entrez_Gene_Id # put gene id in rowname
exp <- exp[c(-1,-2)] # remove columnes of gene id and hugo_symbol
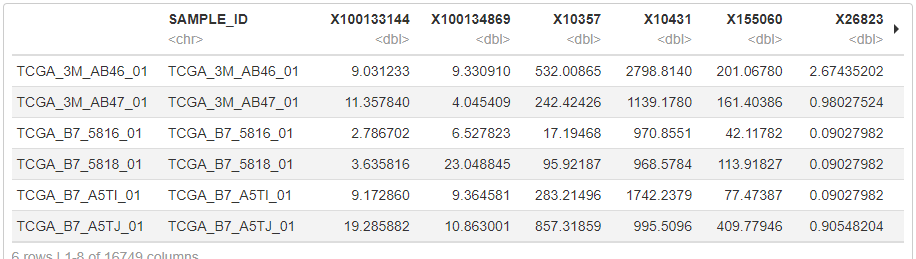
exp <- as.data.frame(t(exp)) # 転置して行と列を入れ替える
# SAMPLE_IDをカラム名として追加する
exp <- data.frame(SAMPLE_ID=rownames(exp), exp)
head(exp)以下のように、SAMPLE_ID, geneID1、geneID2、という形でdataframeを作成できました。
臨床情報準備
# サンプルのクリニカル情報読み込む。skipで不要な始め4行を飛ばす。
clinical <- read.delim("data/stad_tcga_pan_can_atlas_2018/stad_tcga_pan_can_atlas_2018/data_clinical_sample.txt", skip=4)
# 今回は腫瘍タイプと腫瘍採取施設情報を使用するので、これらの情報だけを抜き出します
clinical <- clinical[c("SAMPLE_ID", "TUMOR_TYPE", "TISSUE_SOURCE_SITE")]
# 後ほど、SAMPLE_IDをKeyとしてデータフレームを結合するため、SAMPLE_IDを発現データと同じ表記にする
clinical$SAMPLE_ID <- gsub("-", "_", clinical$SAMPLE_ID)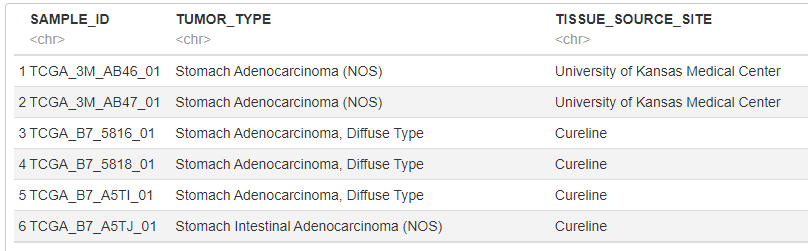
データフレーム結合
臨床情報と発現値情報をSAMPLE_IDをキーとして結合します。
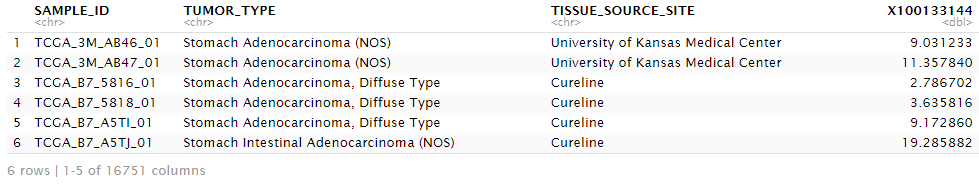
# Merge
d <- merge(clinical, exp, by="SAMPLE_ID")左端にキーとして使用したSAMPLE_IDがあり、次にclinical dataframe由来の列情報、exp dataframe由来の列情報として格納されています。すなわち発現値は4例目からになります。
主成分分析PCAを実施
当たり前なのですが、PCAは数値情報だけをインプットとして受けるとるので、データ指定時に数値情報以外は除いて、インプットします。
# 発現行列だけを使ってPCAを実施 (カテゴリーカラムはすべて除く)
d.pca <- prcomp(d[, c(-1,-2,-3)], scale = T)主成分の固有値(eigenvalue)
各主成分が全体のvarianceのどの程度を説明できるかを示す値が固有値です。簡単にバーチャート表示には”factoextra”を使います。
library("factoextra")
fviz_eig(d.pca, addlabels = TRUE)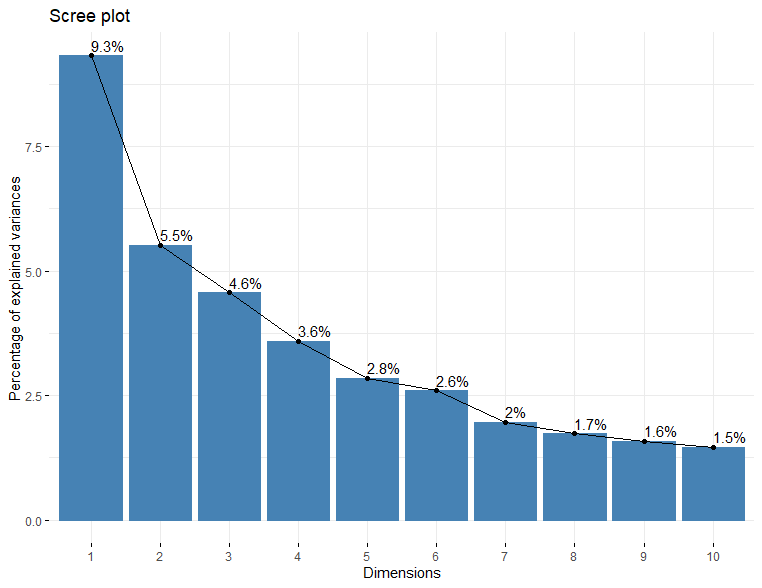
PC1でも全体varianceの9%しか説明できないので、多様性に富んだデータであることが示唆されています。
PCAプロット1
PCAプロットを簡単に表示する方法の1つはfviz_pca_ind()を使用することです。腫瘍採取施設ごとにどのようにデータが分布しているか見てみます。
fviz_pca_ind(d.pca,
col.ind = d$TISSUE_SOURCE_SITE, # color by groups
addEllipses = TRUE, # Concentration ellipses
ellipse.type = "confidence",
legend.title = "TUMOR_TYPE")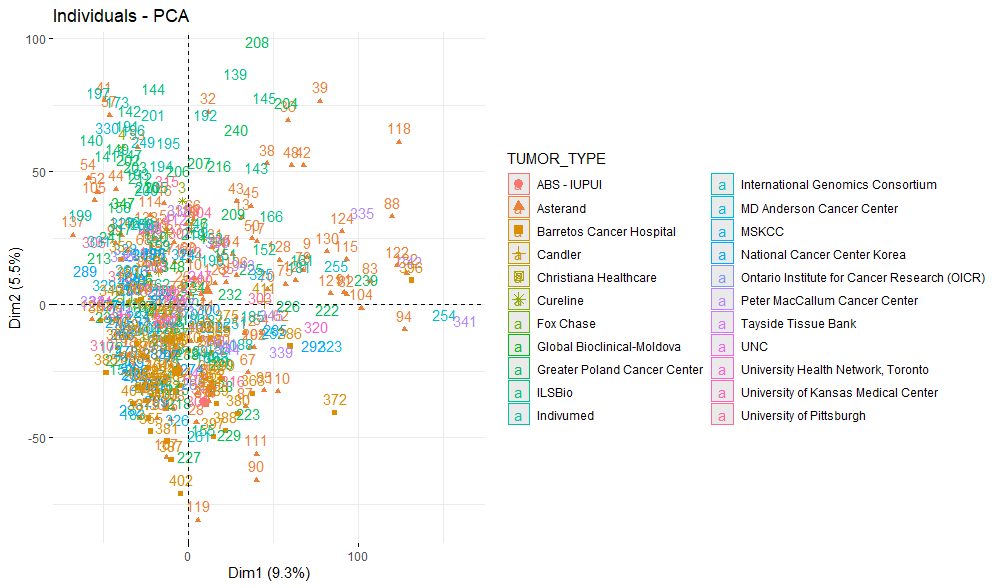
大きな偏りなく各施設由来のサンプルが混在してプロットされているので、サンプル採取施設由来の大きなバイアスはなさそうです。
PCAプロット2
もう少しプロットを自由に編集したいので、次にggplotでプロットを作成してみます。
# PCA結果オブジェクトのx内に各PCが格納されている
PC_score <- as.data.frame(d.pca$x)
library(tidyverse)
ggplot(PC_score, aes(x=PC1, y=PC2, color=d$TISSUE_SOURCE_SITE)) + geom_point()+ theme_bw()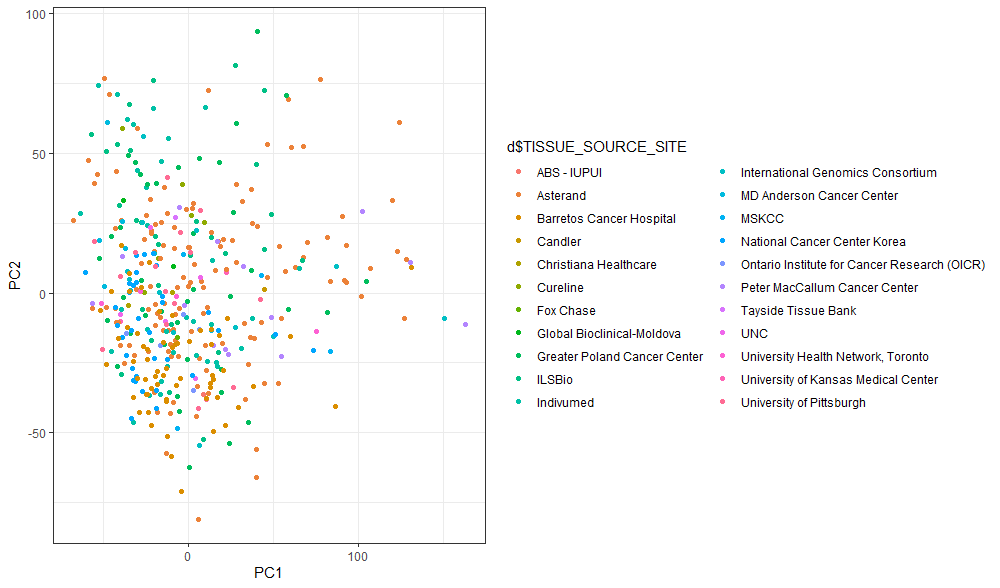
こちらのほうが、図とレジェンドがすっきりしていて、データを見やすいのではないかと思います。
まとめ
以上のように、トランスレーショナルリサーチにおいて、解析を始める前に決まってしまっている因子(施設、サンプル処理時間など)の影響が、どの程度測定データに影響を与えているかどうかの確認は非常に重要なポイントで、研究の信頼性を担保するためにも、必ず自分の目で確認しなければなりません。このステップは研究のスタートポイントになるようなデータに対して、一番初めに実施するべき事前解析であり、いつでもすぐに実行できるようになっておく必要があると筆者は思っています。
今回も最後までお読みいただきありがとうございました。

